
The Condensed Matter Dynamics group is led by Prof. Andrea Cavalleri and focuses on non-equilibrium phenomena in quantum materials. In particular, we investigate how electronic and structural order can emerge as a result of external drives and how transitions occur in complex solids dynamically. Light-control of superconductivity, ferroelectricity, magnetism and topology have been at the centre of our research.
Recent Research Highlights



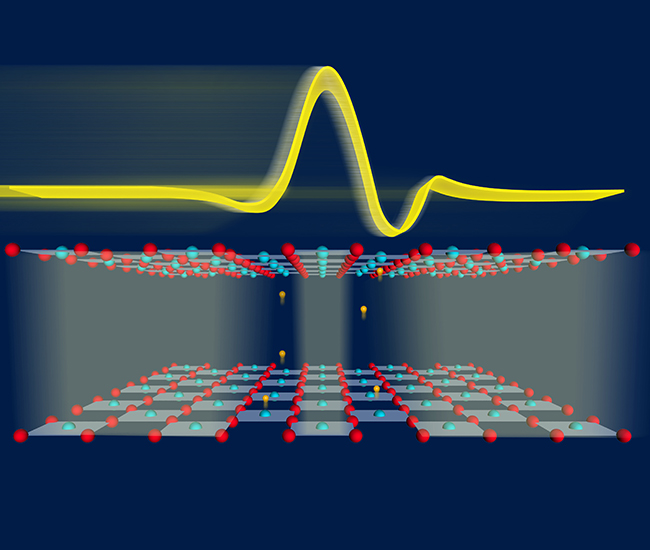

Nonlinear Phononics
Nonlocal nonlinear phononics
M. Henstridge et. al.,Nature Physics, 18, pages 457–461 (2022)
Probing the interatomic potential of solids with strong-field nonlinear phononics
A. von Hoegen et. al., Nature, 555, 79–82 (2018)
Nonlinear lattice dynamics as a basis for enhanced superconductivity in YBa2Cu3O6.5
R. Mankowsky et. al., Nature 516 , 71–73 (2014)
more ...
Nonlinear Phononics
Nonlocal nonlinear phononics
M. Henstridge et. al.,Nature Physics, 18, pages 457–461 (2022)
Probing the interatomic potential of solids with strong-field nonlinear phononics
A. von Hoegen et. al., Nature, 555, 79–82 (2018)
Nonlinear lattice dynamics as a basis for enhanced superconductivity in YBa2Cu3O6.5
R. Mankowsky et. al., Nature 516 , 71–73 (2014)
more ...
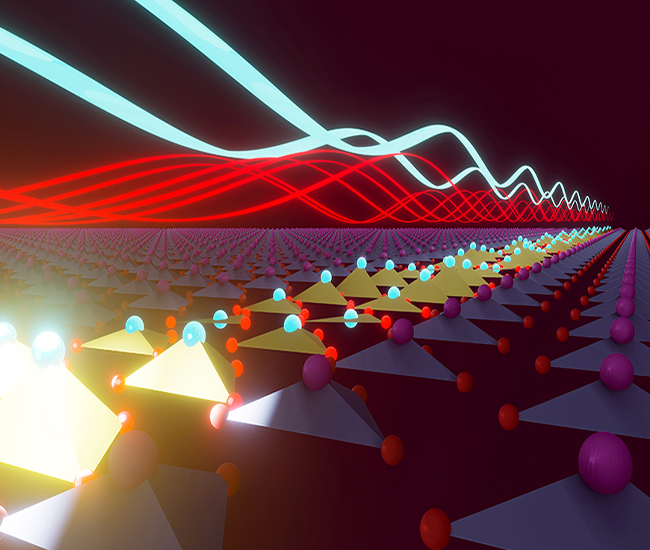
Optical Control of Ferroelectricity
Quenched lattice fluctuations in optically driven SrTiO3
M. Fechner et. al., Nature Materials, 23, 363–368 (2024)
Metastable ferroelectricity in optically strained SrTiO3
T. Nova et. al., Science, 364, 6445, 1075-1079 (2019)
Ultrafast reversal of the ferroelectric polarization
R. Mankowsky et. al., Physical Review Letters, 118, 197601 (2017)
more ...
Optical Control of Ferroelectricity
Quenched lattice fluctuations in optically driven SrTiO3
M. Fechner et. al., Nature Materials, 23, 363–368 (2024)
Metastable ferroelectricity in optically strained SrTiO3
T. Nova et. al., Science, 364, 6445, 1075-1079 (2019)
Ultrafast reversal of the ferroelectric polarization
R. Mankowsky et. al., Physical Review Letters, 118, 197601 (2017)
more ...
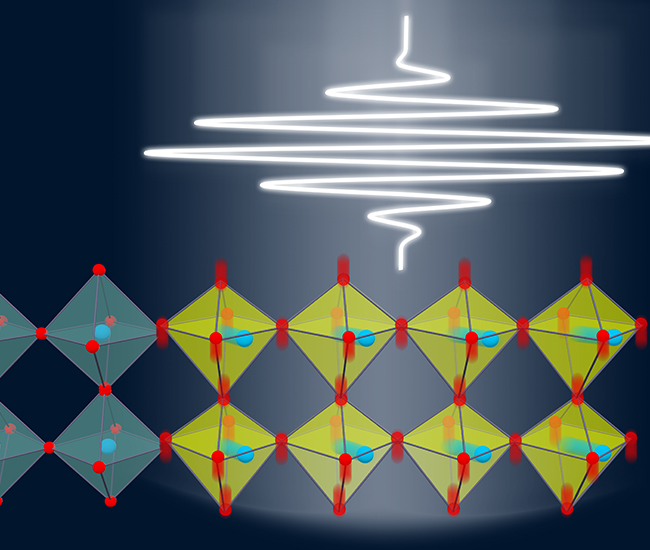
Light-Induced Topological States
Light-induced anomalous Hall effect in graphene
J. W. McIver et. al., Nature Physics,16, 38–41( 2020)
Light-Induced Topological States
Light-induced anomalous Hall effect in graphene
J. W. McIver et. al., Nature Physics,16, 38–41( 2020)
